
January 10, 2023
Im Mai 2022 machte Taysha Gene Therapies die aufregende Ankündigung, dass sie eine klinische Studie der Phase 1/2 zur Gentherapie des Rett-Syndroms beginnen werden. Ihr Plan, MeCP2 im Nervensystem wiederherzustellen, indem sie das funktionelle Protein in Zellen einführen, denen es fehlte, war eine willkommene Entwicklung im Kampf um eine Rett-Heilung. Kurz darauf kündigte Neurogene an, dass sie ebenfalls eine Gentherapie für das Rett-Syndrom erproben wollen. Bei dieser Art des Gen-„Ersatzes“ wird ein Virus eingesetzt, um ein funktionsfähiges MECP2-Gen in das Gehirn einzuschleusen. Eine Besonderheit der Taysha-Studie besteht darin, dass nicht die gesamte kodierende MeCP2-Sequenz verwendet wird, sondern eine gekürzte Version, die viel kürzer ist. Dieser Artikel erklärt, wie das „Mini-MeCP2“ zustande kam. (Hinweis: Ich habe nichts mit Taysha oder der vorgeschlagenen Studie zu tun).
Als meine Forschungsgruppe auf die Idee mit dem Mini-MeCP2 kam, dachten wir nicht in erster Linie an einen therapeutischen Vektor, sondern eher daran, herauszufinden, wie das MeCP2-Protein tatsächlich funktioniert. Alle menschlichen Proteine haben sich so entwickelt, dass sie Funktionen erfüllen, die zu einem gesunden Individuum beitragen. MeCP2 ist da keine Ausnahme, aber es war nicht einfach, herauszufinden, was es genau tut. Verlockende Hinweise ergaben sich aus der Betrachtung der Mutationstypen, die die Krankheit verursachen. Zum Hintergrund: Proteine bestehen aus Ketten von Aminosäuren, von denen es 20 verschiedene Arten gibt. Damit das Protein richtig funktioniert, müssen die richtigen Aminosäuren in genau der richtigen Reihenfolge angeordnet sein. Mutationen bringen diese Reihenfolge durcheinander, entweder auf diskrete Weise, indem sie eine Aminosäure in der Kette durch die falsche ersetzen, oder auf drastische Weise, indem sie den Verlust von Teilen der Kette verursachen. Unabhängig davon, ob es sich um diskrete oder drastische Mutationen handelt, ist das Endergebnis dasselbe: ein ernsthafter Mangel an funktionierendem MeCP2, der zum Rett-Syndrom führt.
Kein Protein agiert allein, daher besteht eine Möglichkeit, die Funktion zu verstehen, darin, nach Partnern zu suchen, die mit ihm zusammenarbeiten. Im Fall von MeCP2 wussten wir schon lange, dass einer seiner Partner die DNA ist – insbesondere methylierte DNA. Im Jahr 2013 fand Matt Lyst, Mitglied des Labors, einen zweiten wichtigen Partner: einen riesigen Multiproteinkomplex namens NCoR. Mithilfe der Biochemie konnten wir diese beiden Interaktionsdomänen auf einzelne Regionen der MeCP2-Proteinkette eingrenzen, die wir MBD (Methyl-CpG Binding Domain) bzw. NID (NCoR Interaction Domain) nannten. Entscheidend ist, dass die große Mehrheit der Rett-Mutationen eine oder beide dieser Domänen inaktiviert. Besonders aufschlussreich waren die subtilen Rett-verursachenden Mutationen, die auf Aminosäuren in der Kette hinweisen, die überhaupt nicht verändert werden können. Unsere Ergebnisse legten eine einfache Hypothese nahe, um zu erklären, was MeCP2 tut: Es dient als Brücke zwischen DNA-Methylierungsstellen im Genom und dem NCoR-Corepressor. Mit anderen Worten: Es rekrutiert NCoR an die genomische DNA von Gehirnzellen – insbesondere von Neuronen, in denen MeCP2 besonders häufig vorkommt. Mutationen, die eines der beiden Enden der MeCP2-Brücke unterbrechen, verhindern die Rekrutierung, was zum Rett-Syndrom führt (siehe Cartoon unten).
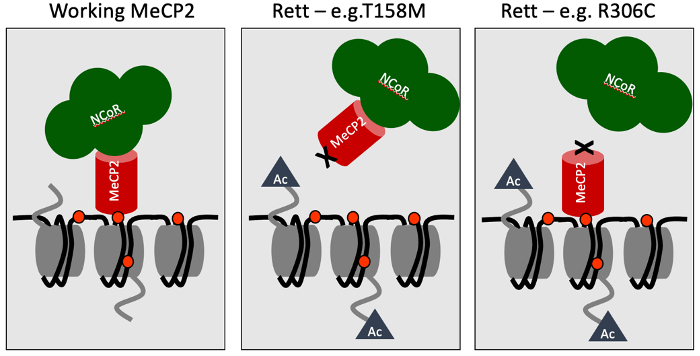
Das „Brückenmodell“ für die Funktionsweise von MeCP2. MeCP2 verbindet spezielle Stellen (rote Punkte) auf der DNA (schwarze Schnur) mit dem NCoR-Komplex (grüne Wolke). Mutationen, die das Rett-Syndrom verursachen, unterbrechen das eine oder andere Ende (oder beide) der Brücke. Die mit „Ac“ gekennzeichneten Dreiecke stellen Acetylgruppen dar, die NCoR entfernt, wenn es in die Nähe des Genoms gebracht wird, wodurch die Ablesung der DNA für die Herstellung von Proteinen verändert wird.
Wissenschaftliche Hypothesen sind im besten Fall wissenschaftliche Einwegwerkzeuge, die zu Experimenten anregen, die das Potenzial haben, das Gegenteil zu beweisen. Die Brückenhypothese sagt beispielsweise voraus, dass sich alle Mutationen beim Rett-Syndrom auf die MBD oder die NID auswirken sollten, aber tatsächlich liegen einige Rett-Mutationen nicht in der Nähe der MBD oder der NID, was darauf hindeutet, dass etwas anderes vor sich gehen könnte. Als Jacky Guy, ein Mitglied meines Labors, diese Mutationen genauer untersuchte, stellte sich jedoch heraus, dass sie dazu führten, dass das mutierte MeCP2-Protein instabil war und folglich abgebaut wurde. Obwohl sich diese Mutationen also nicht direkt auf die MBD oder die NID auswirkten, führten sie zu einem fast vollständigen Verlust von MeCP2. Die Brückenhypothese überlebte diese und andere Tests, aber wir suchten immer noch nach einem „Killer“-Experiment, das das Potenzial hatte, die von uns favorisierte Erklärung für die Funktion von MeCP2 zunichte zu machen.
Die Idee war einfach: Das natürliche MECP2-Gen in Mäusen sollte durch eine verkürzte Version ersetzt werden, die die MBD und die NID beibehält, der aber die restlichen zwei Drittel des Proteins fehlen. Wenn wir richtig lagen, sollten diese beiden Domänen allein in der Lage sein, die Aufgabe von MeCP2 in voller Länge zu erfüllen. Der Grund, warum dieses Experiment riskant erschien, ist, dass die Aminosäuresequenz des MeCP2-Proteins im Laufe der Evolution sehr konserviert ist. Das heißt, die Kette ist bei allen Säugetieren und sogar bei Fischen und Fröschen praktisch gleich geblieben. Da ständig Mutationen auftreten, könnte man erwarten, dass im Laufe der vielen Millionen Jahre der Evolution, die z. B. Mäuse und Menschen trennen, die Sequenz der Aminosäurenkette driften würde, wie es bei vielen anderen Proteinen der Fall ist. Die Tatsache, dass dies bei MeCP2 nicht der Fall ist, lässt vermuten, dass die ganze Sache „genau so“ sein muss, sonst funktioniert sie nicht richtig. Welche Hoffnung gab es, dass das Abschneiden des größten Teils eines solchen Proteins (siehe Diagramm unten) mit normalem Leben vereinbar sein würde?
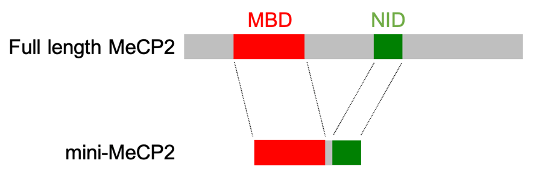
Die Kette mit fast 500 Aminosäuren in voller Länge ist durch einen Balken dargestellt, der die MBD und die NID hervorhebt (siehe Text). Dem von Tillotson und Kollegen erzeugten Mini-MeCP2 fehlen fast 70 % des Proteins in voller Länge, aber die beiden Schlüsseldomänen sind erhalten.
Die Idee stand eine ganze Weile im Raum und wartete auf jemanden, der bereit war, ein risikoreiches Projekt zu übernehmen. Glücklicherweise nahm die Doktorandin Becky Tillotson den Fehdehandschuh auf und zeigte, dass Mäuse, deren MECP2-Gen durch ein für Mini-MeCP2 kodierendes Gen ersetzt worden war, tatsächlich bemerkenswert gesund waren. Ohne MeCP2 sterben die Mäuse einige Monate nach der Geburt, aber die Mini-MeCP2-Mäuse hatten eine Lebenserwartung von mehr als einem Jahr und ähnelten Mäusen ohne Mutation. Jim Selfridge, ein langjähriges Mitglied meines Labors, schlug vor, zu testen, ob das Einschalten des Mini-Gens, nachdem die Mäuse krank geworden waren, ihre Probleme lösen könnte. Dieses Experiment basierte auf den „klassischen“ Umkehrexperimenten von Jacky Guy, die zeigten, dass eine verzögerte Wiederherstellung von MeCP2 die schweren neurologischen Probleme bei Mäusen mit Rett-Syndrom umkehren konnte. Wie sich herausstellte, fand Becky heraus, dass Mini-MeCP2 dasselbe bewirken konnte, was die Ansicht eindrucksvoll untermauert, dass MBD und NID allein das meiste von dem leisten können, was MeCP2 im Gehirn tut. Als Sahnehäubchen arbeiteten wir mit dem Labor von Stuart Cobb zusammen, der damals noch in Glasgow war (inzwischen ist er an der Universität Edinburgh tätig), um herauszufinden, ob die Übertragung des Gens für mini-MeCP2 in einem AAV9-Virus auch Rett-Mäuse retten könnte. Auch hier gab es beeindruckende Verbesserungen, was darauf hindeutet, dass diese Version des Proteins therapeutisch nützlich sein könnte.
Warum sollte man für die Gentherapie eine abgespeckte Version von MeCP2 verwenden und nicht das ganze Ding? Unsere ursprüngliche Studie wies auf zwei potenzielle Vorteile hin. Erstens ist die Nutzlastkapazität von AAV9-Vektoren sehr begrenzt, so dass die Verkürzung des Gens mehr Raum für das Hinzufügen anderer DNA-Bestandteile schafft, um die Menge des Proteinprodukts zu optimieren oder zu regulieren. Zweitens haben wir den Eindruck gewonnen, dass die bekannte Toxizität, die durch zu viel MeCP2 verursacht wird, bei Mini-MeCP2 weniger wahrscheinlich ist. Spätere unveröffentlichte Arbeiten im Labor von Stuart Cobb haben gezeigt, dass zu viel Mini-MeCP2 bei Mäusen ebenfalls toxisch ist, so dass, wie bei MeCP2 in voller Länge, eine übermäßige therapeutische Dosis vermieden werden muss. In beiden geplanten Studien werden Maßnahmen ergriffen, um sicherzustellen, dass die MeCP2-Menge in Grenzen gehalten wird. Trotz der unvermeidlichen Unwägbarkeiten besteht die begründete Hoffnung, dass Mini-MeCP2 einen klinischen Nutzen bringen kann – vorausgesetzt, es kann an genügend Nervenzellen abgegeben werden. Letzteres ist eine große Unbekannte: Ist AAV9 in der Lage, das gesamte Gehirn beim Menschen zu erreichen? Die einhellige Meinung ist, dass wir mit ziemlicher Sicherheit bald einen besseren viralen Vektor brauchen werden, und viele wissenschaftliche und industrielle Labors arbeiten hart daran, einen solchen zu finden. Positiv zu vermerken ist jedoch, dass selbst eine geringe Menge MeCP2 eine deutliche Verbesserung bei Rett-Mäusen bewirkt. Es bleibt zu hoffen, dass die Initiativen von Taysha und/oder Neurogene uns die Ermutigung geben können, nach der wir uns sehnen.